Integrating Cytiva Protein Select tag into an expression vector
Explore guidelines and recommendations for the design and generation of DNA constructs to generate a recombinant protein with the Cytiva Protein Select resin
18 Dec 2024
Cytiva™ Protein Select™ resin is an affinity chromatography resin for purifying recombinant proteins using the self-cleaving Cytiva™ Protein Select™ tag. During an affinity step performed with Cytiva™ Protein Select™ resin, the protein self-cleaves from the tag and elutes with no residual tag amino acids.
To generate a recombinant protein with the Cytiva™ Protein Select™ resin it is essential to design a DNA construct (e.g., a plasmid DNA) that positions the Cytiva™ Protein Select™ tag at the N-terminus of the protein. This article offers guidelines and recommendations for the design and generation of such constructs.
In the era of synthetic biology, genes can be easily and directly synthesized and integrated into a suitable expression vector by several cloning service providers. This permits design of tagged proteins without the need for specific tag vectors. In addition, this approach also eliminates the presence of cloning scars and redundant sequences in the expressed protein, which are commonly seen when the tag is integrated into expression vectors containing tag sequences.
If you intend to conduct the cloning in-house, the following examples delineate two distinct methodologies for generating expression plasmids that correctly integrate the Cytiva™ Protein Select™ tag sequence, ensuring expression of a protein devoid of redundant amino acids at its N-terminus that may arise from cloning scars. While the examples outlined here are specific to generate E. coli expression plasmids with a pET28a expression vector backbone (MilliporeSigma™, cat. no. 69864-3), the cloning principles described are applicable for generating other expression plasmids suitable for various expression host cells.
Method 1: Restriction enzyme-based cloning
Restriction enzyme (RE) cloning is the traditional way of cloning and, for many reasons, remains one of the most popular techniques. In brief, RE cloning involves cutting an expression vector and a linear DNA fragment (insert or gene) at specific sites. This allows joining of the fragment and expression vector by a DNA ligase, generating a complete circular recombinant expression plasmid that can be used as a template for protein expression.
To demonstrate the introduction of the Cytiva™ Protein Select™ tag, the DNA sequence for mature interleukin-1 beta (IL-1β) was amplified using polymerase chain reaction (PCR). PCR primers were designed with overhangs to introduce the tag sequence upstream of IL-1β and to ensure cloning compatibility with the expression vector. More specifically, the PCR primer design used introduces suitable RE sites to ensure compatibility with traditional RE cloning and ligation to the pET28a vector. Overlapping expression vector sequences were also introduced by the PCR primer design to ensure compatibility when performing Gibson assembly cloning with the pET28a vector (Fig 1). The 5’ overhangs may be significantly reduced in length, and do not need to be destination vector compatible if the aim is to only use restriction enzyme-based cloning. The overhangs should be long enough (required length depends on the RE) and in many cases there are no specific sequence requirements.
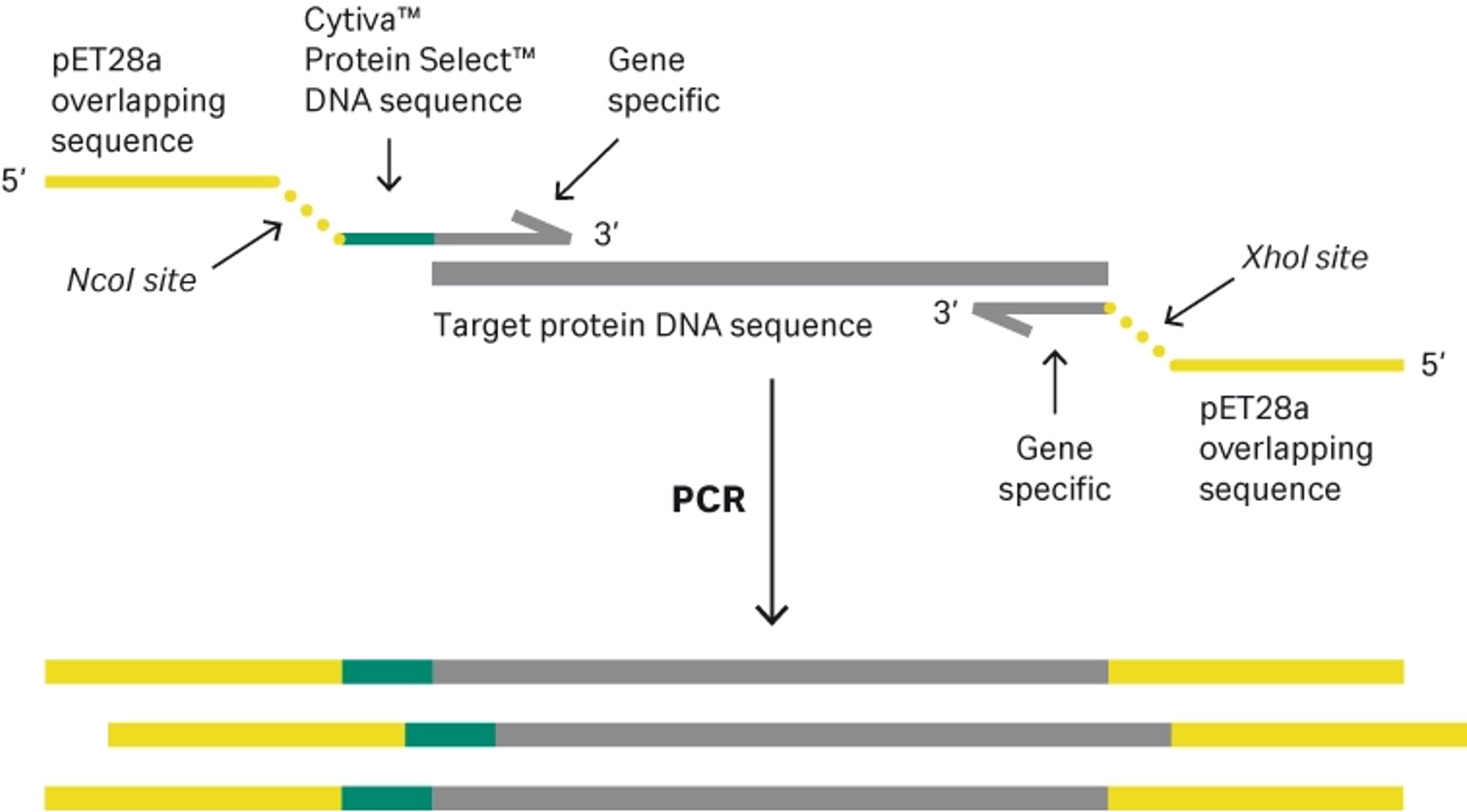
Fig 1. Primer design compatible with both restriction enzyme-based cloning and Gibson assembly.
The forward primer is also designed so that a proline to phenylalanine mutation at the second amino acid position (P2F) is introduced in the IL-1β sequence during PCR amplification—the amino acid proline is undesirable at the N-terminus because it impairs cleavage kinetics as described in FAQs about recombinant protein purification - cleavage mechanisms. A standard 35-cycle PCR was performed with a proofreading DNA polymerase to amplify the mature IL-1β DNA sequence, and the PCR reactions were subjected to agarose gel electrophoresis.
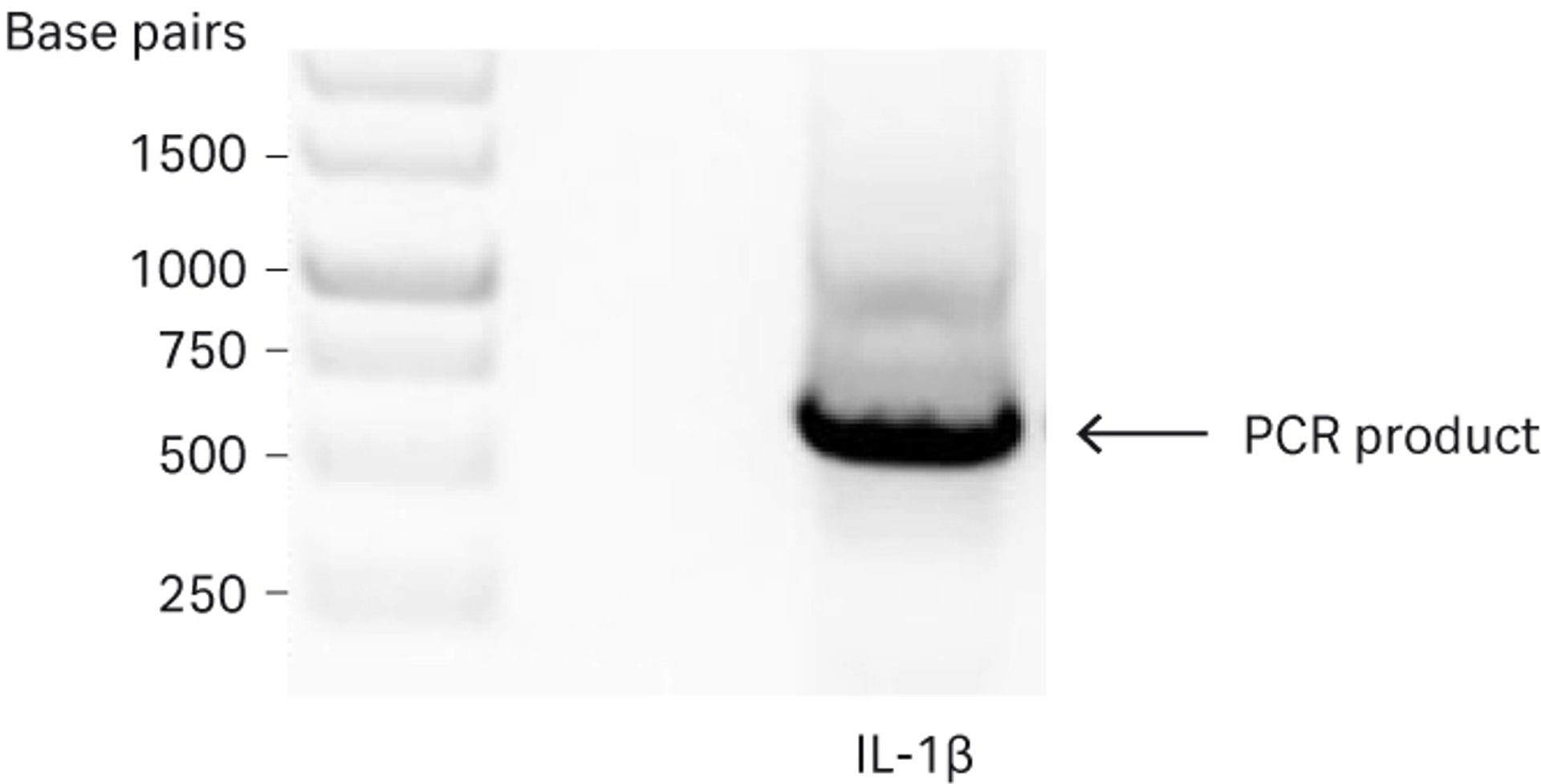
Fig 2. Agarose gel analysis of the PCR amplified mature IL-1β sequence. Amplified IL-1β is indicated by the arrow.
The purified PCR product and pET28a vector were digested with NcoI and XhoI according to the manufacturer’s instructions (Fig 3).
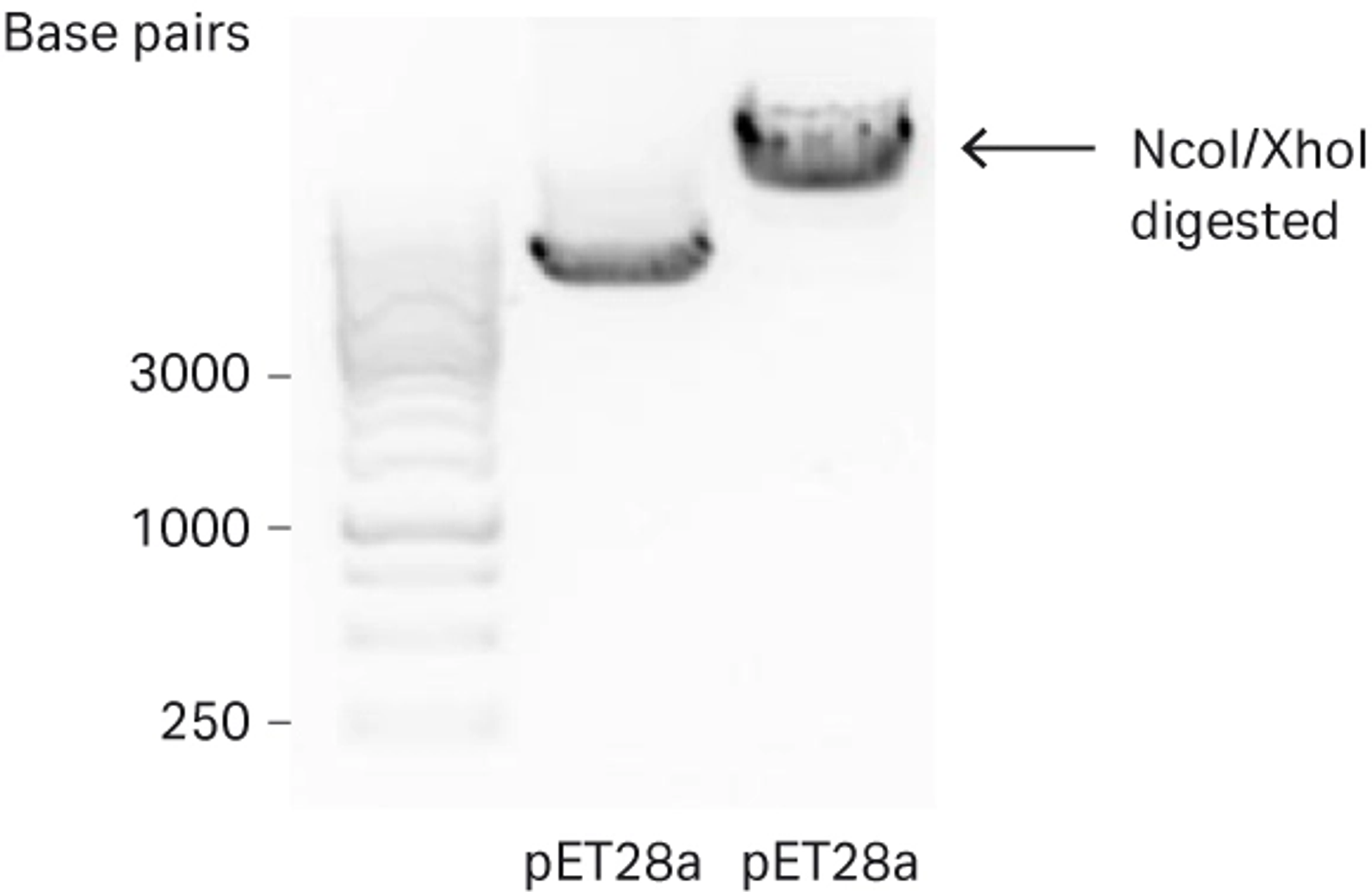
Fig 3. Agarose gel analysis of pET28a vector digested with NcoI and XhoI used for further downstream cloning.
The pET28a vector was additionally treated with alkaline phosphatase to reduce risk of re-ligation of the vector and subjected to agarose gel electrophoresis. Both the linearized vector and digested PCR product were purified with the GFX PCR DNA and Gel Band Purification Kit. Concentrations of the purified vector and PCR-generated IL-1β insert were determined by UV absorbance at 260 nm. The ligation reaction setup used a molar 3:1 insert to vector ratio and T4 DNA ligase, in accordance with the manufacturer’s instructions.
The ligation reaction was incubated for 3 h at room temperature and 2 µL of the reaction was used to transform 50 µL of TOP10 competent cells according to the manufacturer’s instructions, before spreading on Luria-Bertani (LB) agar plates supplemented with the appropriate selection antibiotic. Positive clones, containing the recombinant plasmid, were cultivated with the appropriate antibiotic in LB media and plasmid was subsequently purified using a PlasmidPrep Mini Spin Kit. Correct sequences were confirmed by Sanger sequencing.
Method 2: Gibson assembly technique
Gibson assembly is a cloning technique that allows the joining of two or more DNA fragments in a single isothermal reaction and is driven by three enzymatic reactions. This technique circumvents the need for restriction enzymes and subsequent purifications if the insert and expression vector are already prepared for this purpose (i.e., enabled by the PCR primer design that introduced expression vector homologous sequences during PCR amplification of the insert). Once the reaction is completed, the recombinant plasmid is then ready to be transformed into a suitable expression host cell system.
As illustrated in Figure 1, the 5’ overhangs of the primers were designed to ensure compatibility with the Gibson assembly technique. Undigested but purified PCR fragments and the NcoI and XhoI digested pET28a vector were combined. After a 30 min isothermal incubation at 50°C, the assembled mixture was transformed as stated earlier and plated on LB agar plates supplemented with the appropriate selection antibiotic. Colonies were picked, cultivated and pDNA was then isolated and sent for Sanger sequencing to confirm correct and identical DNA sequences as the restriction enzyme digest strategy.
Confirming protein expression
Once an expression construct has been generated, and the correct DNA sequence is verified, the construct may be transformed into an expression host cell to evaluate protein expression.
The correct and sequence verified pDNA was transformed into an E. coli BL21(DE3) expression strain. Two random clones were picked and inoculated in 5 mL overnight LB culture. The overnight culture was then diluted 1:100 in LB media and cultivated until optical density at 600 nm (OD600) reached 0.7. Small aliquots of both cultures were harvested to use as pre-induction control samples. The temperature of the incubator was reduced to 22°C and expression was induced overnight by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) (final concentration 0.5 mM). The next day, small aliquots of both cultures were collected to use as induced samples. The remaining overnight expression cultures were harvested by centrifugation at 5000 RCF (relative centrifugal force) for 30 min and pellets were stored at -20°C until use. Protein expression of the induced samples and pre-induction control samples was evaluated by SDS-PAGE and subsequent visualization of proteins was performed by Coomassie staining (Fig 4).
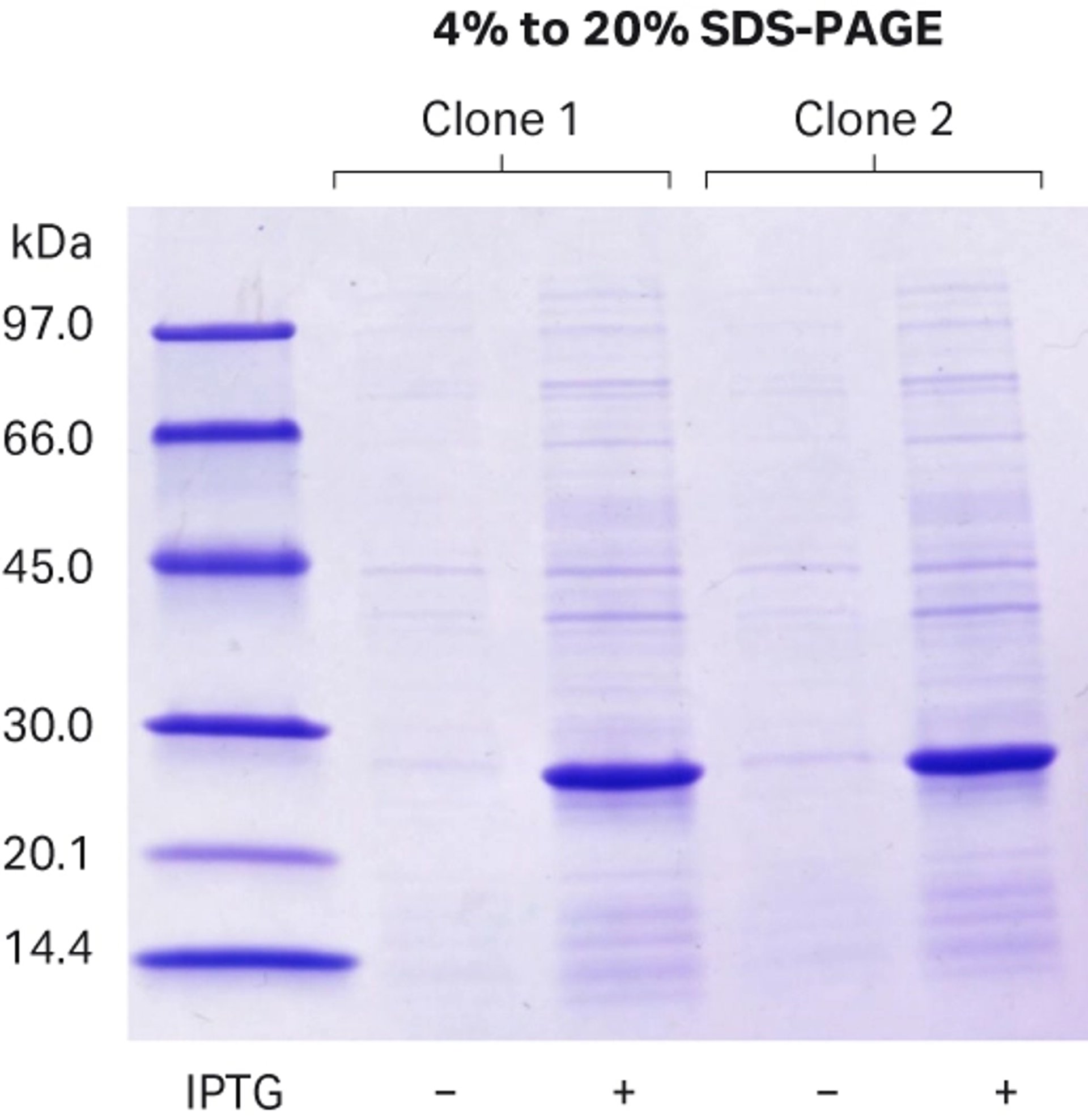
Fig 4. SDS-PAGE analysis indicates expression of the tagged target protein at expected molecular weight (22.4 kDa).
Primer sequences
Forward primer enabling integration of tag protein into an expression vector
Figure 5 illustrates the design of forward primer that enables integration of Cytiva™ Protein Select™ tag into a pET28a expression vector.
The primer overlapping pET28a sequence is a 39 nucleotide sequence. It has the following 5’-3’ sequence: AATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGG.
- The start codon of Cytiva™ Protein Select™ tag sequence is identified in bold text.
- The NcoI site is underlined.
- The sequence for the Cytiva™ Protein Select™ tag and target protein DNA sequence must be included for desired PCR amplification.
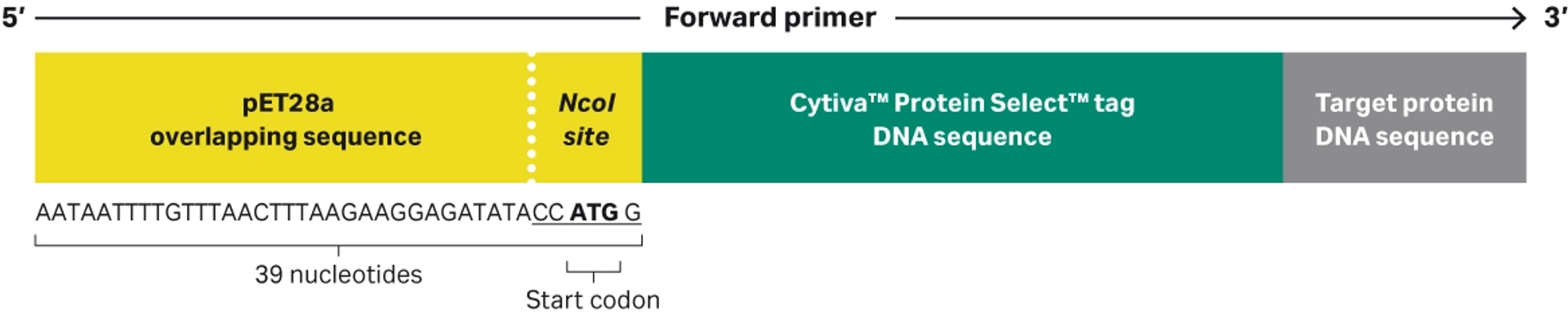
Fig 5. Illustration of the forward primer including the restriction site and pET28a overlapping sequence that allows for integration by RE cloning and Gibson assembly.
Reverse primer enabling integration of an amplified DNA sequence into an expression vector
Figure 6 illustrates the reverse primer that enables integration of the amplified DNA sequence into a pET28a expression vector and allows expression of the optional vector-derived C-terminal his-tag, that in this example was included for detection purposes.
The pET28 overlapping primer sequence is a 27 nucleotide sequence. It has the following 5’-3’ sequence: TCAGTGGTGGTGGTGGTGGTGCTCGAG.
- The Xhol site is underlined.
- The opal stop codon is identified in bold text.
- A reverse complementary sequence for the target protein DNA sequence must be included for desired PCR amplification.
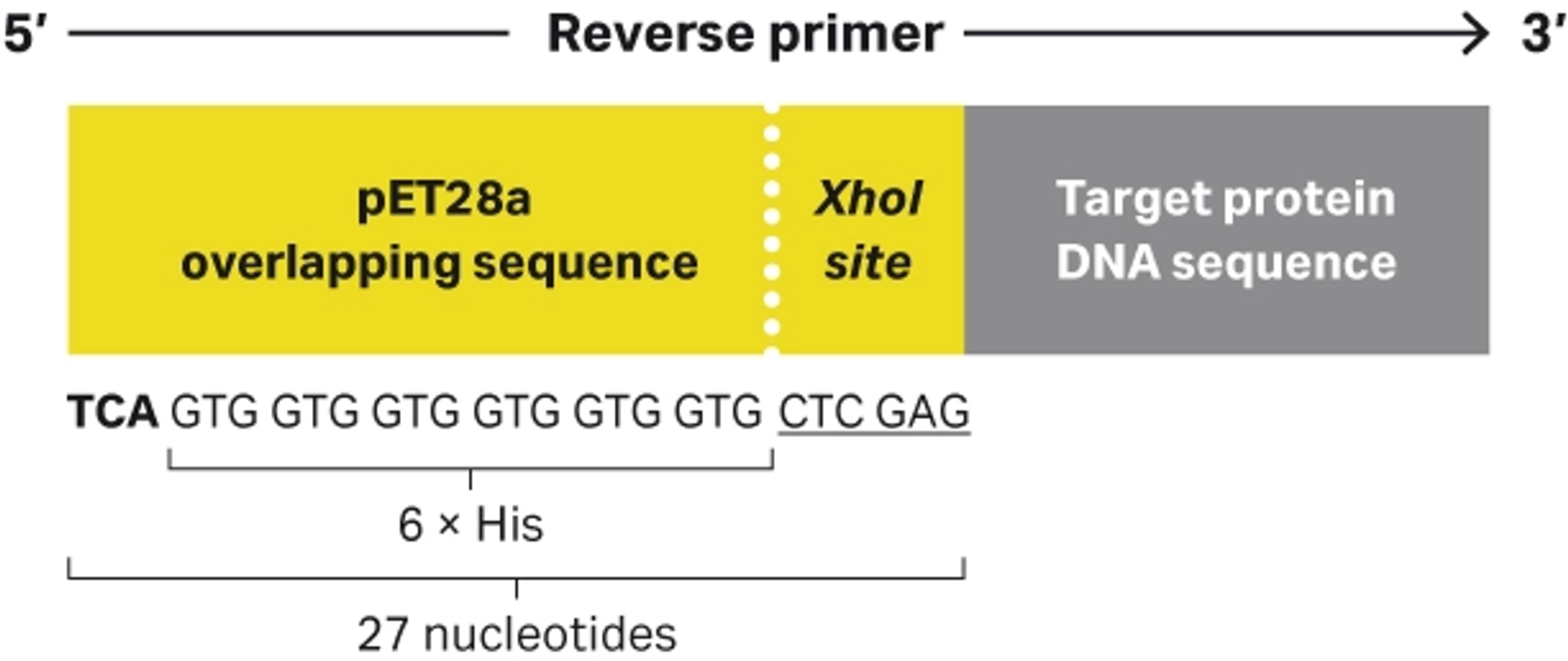
Fig 6. Illustration of the reverse PCR primer including the restriction site and pET28a overlapping sequence that allows for integration by RE cloning and Gibson assembly with a C-terminal 6 × His.
Summary
This article has provided guidelines for designing and generating expression constructs that include Cytiva™ Protein Select™ tag. Such constructs can be generated by a synthetic biology approach or by in-house cloning as outlined by the two methods shown here. Independently of the methods, the experimental design presented incorporates the DNA sequence of the Cytiva™ Protein Select™ tag by PCR. This cloning concept can be used, with some tailored sequence changes, for the majority of available expression vectors.
- Method 1. Restriction-enzyme based cloning
This traditional cloning method involved cutting and joining DNA fragments (e.g., PCR-generated insert and expression vector). Here we amplified a Cytiva™ Protein Select™ tag and IL-1 β DNA sequence and inserted it into a pET28a vector.
- Method 2. Gibson assembly technique
This method joins DNA fragments in a single isothermal reaction without the need for restriction enzymes. The primer design ensured accurate amplification of Cytiva™ Protein Select™ tag and IL-1β DNA sequence and its compatibility with a pET28a vector assembly reaction.
- Confirming protein expression
The sequence verified constructs were propagated, expression was induced, and successful expression was recorded.
*This article was authored by Johan Ohman, Cytiva.
