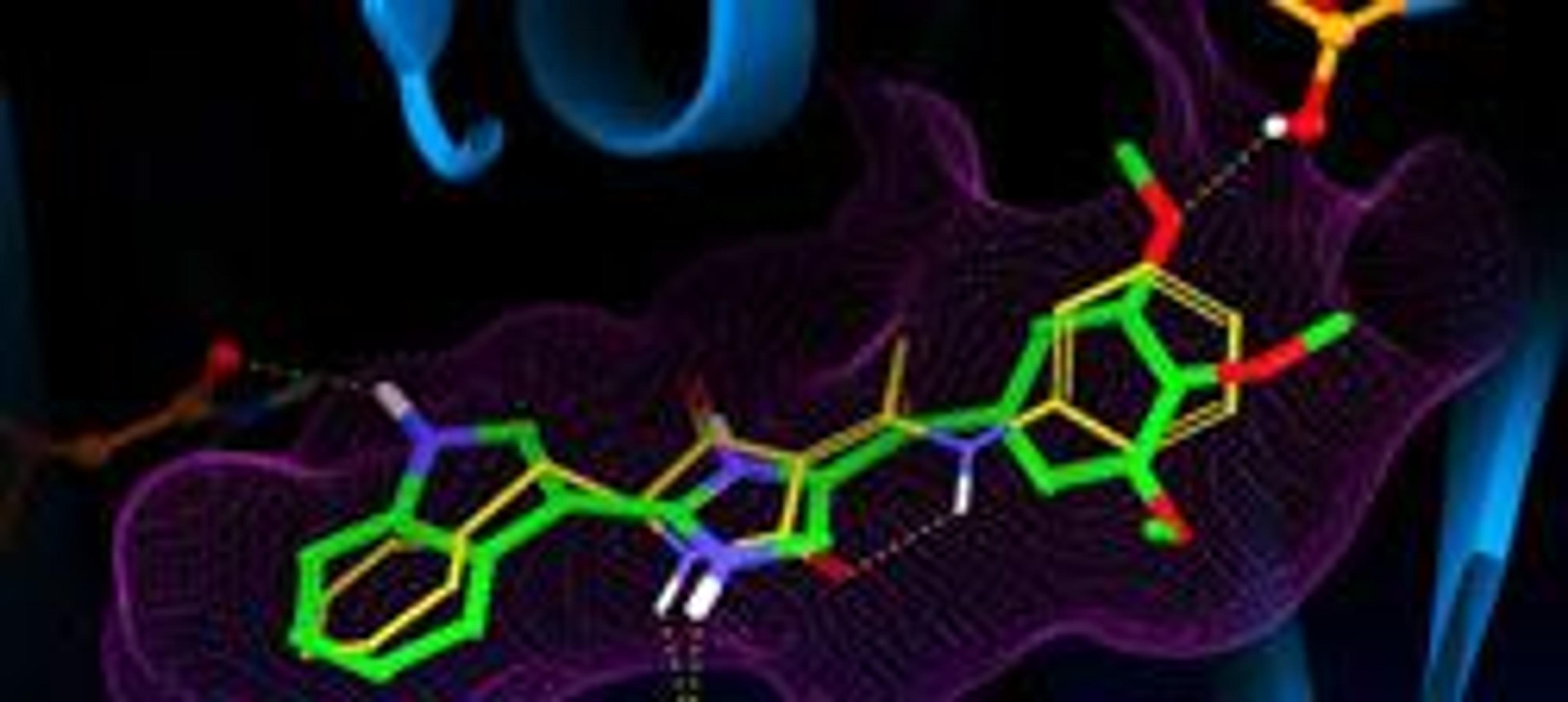
Glide - A complete solution for ligand-receptor docking
Glide: A complete solution for ligand-receptor docking Glide offers the full spectrum of speed and accuracy from high-throughput virtual screening of millions of compounds to extremely accurate binding mode predictions, providing consistently high enrichment at every level. The Advantages of Computational Docking The widespread use of combinatorial chemistry and high-throughput screening (HTS) in the pharmaceutical and biotech…
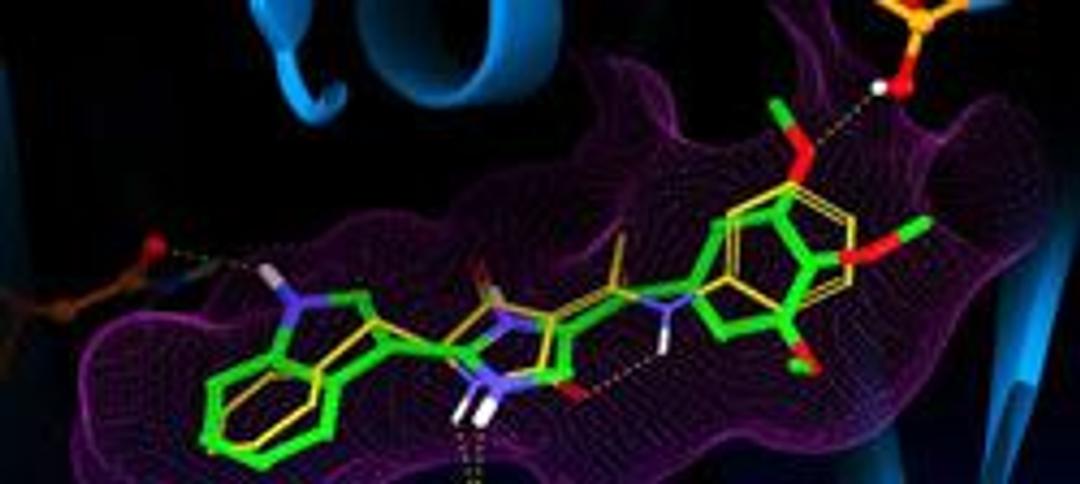
The supplier does not provide quotations for this product through SelectScience. You can search for similar products in our Product Directory.
Best docking software.
Docking
Best docking software, but "feeling the heat" from open source tools.
Review Date: 28 Jan 2020 | Schrödinger
Molecular Modeling
1. Boosting virtual screening enrichment using a new data fusion approach: Coalescing 2D fingerprints, shape, and docking. 2. Progress in pKa prediction with ab initio methods: A new hierarchical model for functional groups. 3. This is the only known software in which work is recognized by noble prize.
Review Date: 28 Feb 2014 | Schrödinger
Molecular Modelling
This is an excellent docking program.
Review Date: 18 Feb 2014 | Schrödinger
Bioinformatics
Best docking software with excellent feasibility in doing high throughput screening.
Review Date: 25 Sept 2013 | Schrödinger
Glide: A complete solution for ligand-receptor docking
Glide offers the full spectrum of speed and accuracy from high-throughput virtual screening of millions of compounds to extremely accurate binding mode predictions, providing consistently high enrichment at every level.
The Advantages of Computational Docking
The widespread use of combinatorial chemistry and high-throughput screening (HTS) in the pharmaceutical and biotechnology industries means that large numbers of compounds can now routinely be investigated for biological activity. However, screening large chemical libraries remains an expensive and time-consuming process, with significant rates of both false positives and false negatives.
High-speed computational methods can now enrich the fraction of suitable lead candidates in a chemical database, thereby creating the potential to greatly enhance productivity and dramatically reduce drug development costs. With an ever increasing number of drug discovery projects having access to high-resolution crystal structures of their targets, high-performance ligand-receptor docking is the clear computational strategy of choice to augment and accelerate structure-based drug design.